
Translate this page into:
A rare presentation of autosomal recessive proximal renal tubular acidosis

*Corresponding author: Aishwarya Ajit Godbole, Department of Ophthalmology, M&J Western Regional Institute of Ophthalmology, Civil Hospital, Ahmedabad, Gujarat, India. aishwaryagodbole7@gmail.com
-
Received: ,
Accepted: ,
How to cite this article: Godbole AA, Mansuri FF, Gaur AN, Bhagat PR. A rare presentation of autosomal recessive proximal renal tubular acidosis. Glob J Cataract Surg Res Ophthalmol. 2023;2:82-5. doi: 10.25259/GJCSRO_22_2023
Abstract
Proximal renal tubular acidosis (pRTA) is characterised by an inability of the kidneys to reabsorb bicarbonate from the proximal convoluted tubule leading to urinary loss of bicarbonate ions causing metabolic acidosis. It can present as an isolated pRTA or be associated with Fanconi syndrome. Isolated inherited pRTA is exceedingly rare. The inheritance can be autosomal dominant, autosomal recessive (AR), or sporadic. The AR form is linked to a SLC4A4 (NBCe1) mutation and is associated with glaucoma, cataract, band keratopathy, mental and growth retardation requiring lifelong alkali therapy and ocular care. The prognosis is good although extra-renal symptoms may persist and ocular abnormalities may progress with age. Here, we report a rare case of congenital glaucoma associated with cataracts, band keratopathy, growth retardation, and mental retardation in a young female having isolated AR pRTA with SLC4A4 gene mutation.
Keywords
Proximal renal tubular acidosis
Autosomal recessive
SLC4A4
Glaucoma
Band Keratopathy
Metabolic acidosis

INTRODUCTION
Proximal renal tubular acidosis (pRTA) is a disease affecting the proximal tubule function resulting in metabolic acidosis. Inherited isolated pRTA is rare and the age of presentation is variable. The inherited isolated form is more commonly autosomal recessive (AR), with the SLC4A4 gene encoding basolateral sodium-bicarbonate cotransporter (NBCe1). Its mutation leads to reduced activity and/or trafficking, thus disrupting the normal bicarbonate (HCO3-) reabsorption in proximal tubules, leading to pRTA, severe growth retardation, mental retardation, migraine, calcification of basal ganglia, and ocular abnormalities such as glaucoma, band keratopathy, and cataracts.[1-4] NBCe1 found on corneal endothelium transports sodium (Na+) and HCO3 − from the corneal stroma into the aqueous humour, defects which would lead to increased stromal HCO3− facilitating calcium deposition and band-shaped keratopathy formation. It is also thought to play a similar role in the maintenance of lens transparency explaining cataract formation, nevertheless, the role of NBCe1 in the pathogenesis of glaucoma remains unclear.[5] Ocular abnormalities as seen in this patient are typical of inherited isolated pRTA and can prove to be diagnostic clinchers for a disease as rare. Inherited pRTA requires lifelong bicarbonate replacement therapy in large doses. A mixture of sodium and potassium (bicarbonate or citrate) salts may be supplemented to prevent hypokalemia. Thiazide diuretics can also be given to enhance HCO3− reabsorption.[6] Ocular manifestations such as band-shaped keratopathy can be managed by giving topical ethylene diamine tetra acetic acid (EDTA) eye drops and glaucoma can be managed medically by the use of topical intraocular pressure (IOP) lowering drugs and surgically by methods such as trabeculectomy and goniotomy. With proper treatment, the prognosis of acidosis is good, although extra-renal symptoms do not resolve with alkali treatment and ocular abnormalities may show progression with age.[7]
CASE REPORT
An 11-year-old female was clinically diagnosed to have inherited isolated pRTA and presented to our tertiary care centre in March 2023 for ophthalmic evaluation. The patient had come with the genetic study report which confirmed the presence of c.1170G>C; p.R390S of SLC4A4 gene in homozygous state in AR form [Figure 1].
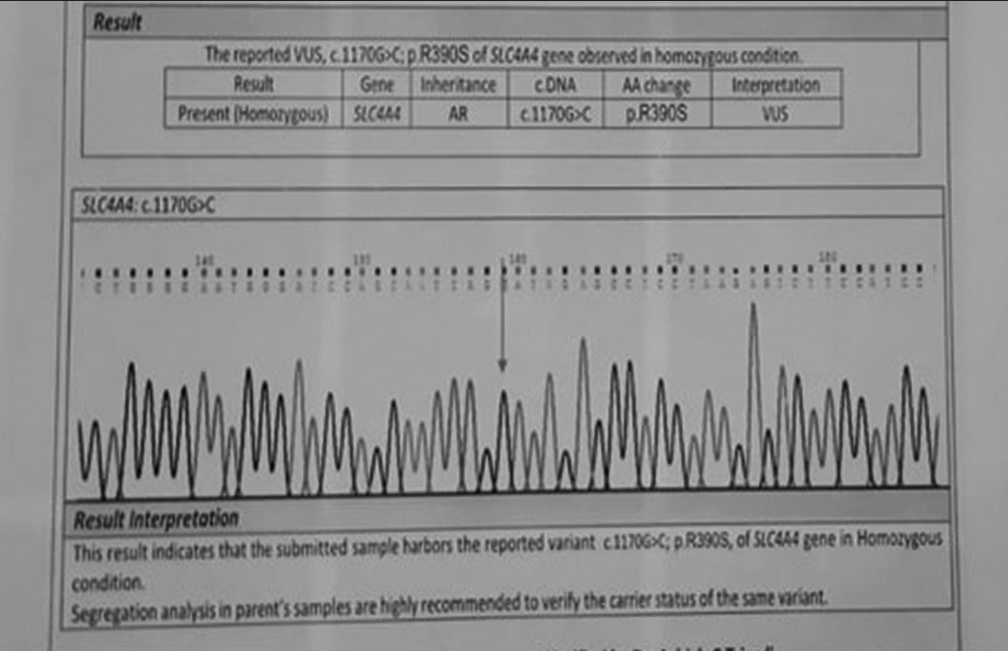
- Genetic study by Sanger sequencing showing the presence of SLC4A4 mutation in homozygous state.
On evaluation, the visual acuity was found to be 6/60, and counting fingers 2 m in the right and the left eye, respectively, did not improve with correction or pinhole. On slit lamp examination, irregular pupils, band-shaped keratopathy changes, and multiple posterior synechiae were noted in both eyes [Figures 2a and b].
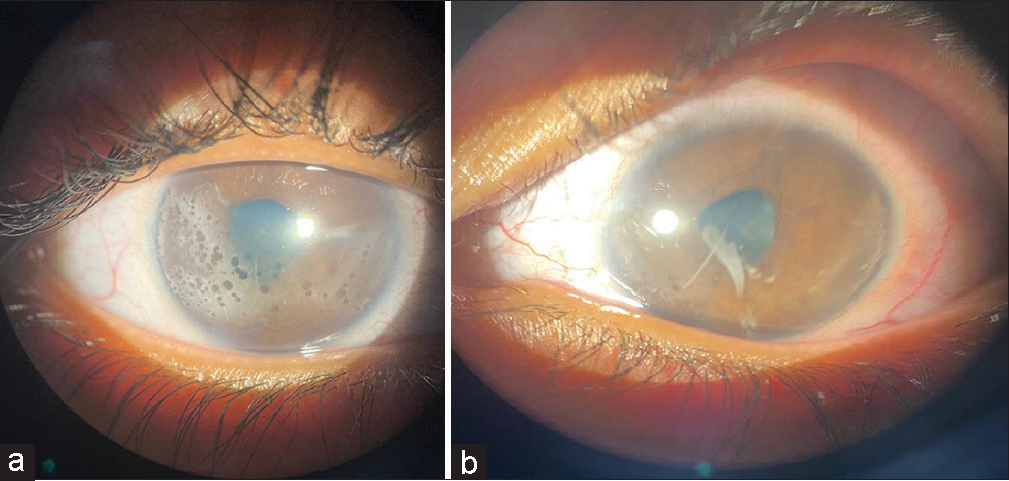
- Slit lamp image of (a) the right eye shows irregular pupil, multiple posterior synechiae, and band-shaped keratopathy changes. (b) The left eye shows irregular pupil, multiple posterior synechiae, and band-shaped keratopathy changes (more in the right eye).
The IOP was measured using Perkin’s handheld applanation tonometer and was found to be 40 and 32 mmHg in the right and the left eye, respectively. The patient was uncooperative for gonioscopy, ultrasound biomicroscopy (UBM), and anterior segment optical coherence tomography (ASOCT). Fundus examination showed an indistinct cup disc ratio of 0.3 and 0.6 in the right and the left eye, respectively, with no other discernible findings.
History
The patient initially presented in February 2020 at 8 years of age with complaints of headache and ocular pain for which the patient had been initially taken to a nearby eye hospital where she was diagnosed to have congenital glaucoma. The patient was the younger of two siblings; the elder sibling was a known case of congenital glaucoma and identified pRTA with SLC4A4 homozygous mutation. There was also a history of glaucoma in the patient’s maternal grandfather, the cause of which was unknown. The IOP was noted to be 32 mmHg in both eyes and the patient was started on topical intraocular pressure lowering drugs, the details of which were not available, and was referred to a higher centre for further work-up. In the local higher centre, the patient was also observed to have growth retardation, mental retardation, metabolic acidosis with a normal anion gap, and normal distal acidification of urine, also there was no evidence of nephrocalcinosis or generalised proximal renal tubular dysfunction. These signs along with the history of the elder sibling led to a clinical diagnosis of inherited isolated pRTA and the patient was advised to undergo a genetic study (hotspot detection by Sanger technique to confirm the presence of SLC4A4 mutation) and referred for detailed ophthalmic evaluation and management at a tertiary centre following which she had presented to our centre.
On evaluation at our centre the visual acuity was documented to be 6/24 in the right eye and counting fingers 2 m in the left eye, the slit lamp showed central lenticular opacity with band-shaped keratopathy changes and the presence of posterior synechiae in both eyes. The patient then was subjected to applanation tonometry with Perkin’s handheld applanation tonometer, gonioscopy, UBM, ASOCT, and fundus examination but the patient was uncooperative for all the above procedures. The patient was started on travoprost 0.004% eye drops once at bedtime, brimonidine 0.1% eye drops twice a day, carboxymethylcellulose 0.5% eye drops 4 times a day and EDTA eye drops 4 times a day in both eyes and advised to undergo an ocular examination under anaesthesia followed by trabeculectomy if required. They were also advised to bring the maternal grandfather and sibling for assessment.
The patient had been lost to follow-up since then and presented again in 2023 but had stopped topical medications. The maternal grandfather and sibling refused to come for any work-up.
At present, the patient had no other systemic symptoms and was compliant with the oral sodium bicarbonate started by the physician. Her blood profile was within normal limits. Electrocardiogram showed normal sinus rhythm and 2D echocardiography revealed the presence of a small ostium secundum atrial septal defect with the left to right shunt, for which no active management was recommended, and a follow-up of 6 months was given by the cardiologist.
The patient was restarted on the prior regimen with the addition of ripasudil 0.4% eye drops twice a day in both eyes and again emphasised for ocular examination under anaesthesia and further management. When the risks associated with general anaesthesia in a patient of pRTA were again explained to the parents, the parents refused the intervention and consented only to topical therapy and follow-up and also refused their own genetic analysis.
DISCUSSION
Inherited pRTA is rare and is commonly AR. The SLC4A4 gene encodes three basolateral sodium-bicarbonate cotransporter (NBCe1) variants: NBCe1-A, NBCe1-B, and NBCe1-C.[6] NBCe1-A mutation leads to pRTA and ocular abnormalities such as band keratopathy, increased corneal opacity, glaucoma, or at least elevated IOP and cataracts.[7-9] NBCe1-B mutation leads to intestinal abnormalities and NBCe1-C mutation leads to neurological abnormalities.[10]
The management of pRTA is alkali therapy to treat acidosis. A Jewish–Georgian woman having normal intelligence, short stature, deformed teeth, and no perception of light had a history of being diagnosed with bilateral glaucoma in early childhood for which she was treated with oral acetazolamide from the age of 6 years and had undergone several operative procedures for control of the high IOP. The patient had developed bilateral cataracts and band keratopathy over the coming years and lost all her vision by the age of 16 years. Metabolic acidosis was first noted when the patient was 33 years of age and she tested positive for a novel mutation of NBCe1 (SLC4A4) with pRTA.[8] Demirci et al. identified a novel, homozygous, missense SLC4A4 mutation in 27 years male Caucasian having pRTA, short stature, enamel hypoplasia, migraine, transient hemiparesis attacks, ataxia, and bilateral ocular manifestations such as cataracts, glaucoma, and band keratopathy.[5] The patient first had disturbances in his vision during his kindergarten years but his eye examination then was completely normal. He later developed progressive myopia, corneal fine granules, and minute lenticular snowflake-type opacities. Gradually, cataracts and band-shaped keratopathy became more evident and he was also diagnosed with bilateral open-angle glaucoma which was initially treated with topical medication but eventually required surgical interventions. EDTA chelation was also advised for the band keratopathy but eventually, the patient lost his eyesight by the age of 27 years.
In both the above cases, the presenting features were ocular abnormalities that progressed with age. The glaucoma was initially treated medically but eventually, surgical treatment was required, despite which both the patients lost their eyesight. This makes counselling of such patients with regard to the visual prognosis of paramount importance.
In our case too, the patient first presented with ocular symptoms, and a similar pattern of progression of ocular symptoms was seen in our patient. Glaucoma in this patient was managed medically as the parents refused any surgical intervention when the visual prognosis, need for multiple surgeries and risk of general anaesthesia was explained. Considering the morbidity of this condition, it is imperative to have regular long-term follow-ups, monitoring of systemic parameters, and aggressive counselling of parents.
CONCLUSION
Ocular manifestations such as glaucoma, band keratopathy, and cataracts may be the presenting features of rare familial disorders such as inherited AR pRTA. Prompt recognition of ocular and systemic features can lead to early diagnosis and can provide an opportunity for efficient treatment. A multidisciplinary approach and parental counselling are also vital for overall management.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent.
Conflicts of interest
Dr.Purvi Raj Bhagat is on the editorial board of the Journal.
Use of artificial intelligence (AI)-assisted technology for manuscript preparation
The author(s) confirms that there was no use of artificial intelligence (AI)-assisted technology for assisting in the writing or editing of the manuscript and no images were manipulated using AI.
Financial support and sponsorship
Nil.
References
- Renal tubular acidosis: Developments in our understanding of the molecular basis. Int J Biochem Cell Biol. 2005;37:1151-61.
- [CrossRef] [PubMed] [Google Scholar]
- Unraveling the molecular pathogenesis of isolated proximal renal tubular acidosis. J Am Soc Nephrol. 2002;13:2171-7.
- [CrossRef] [PubMed] [Google Scholar]
- The divergence, actions, roles, and relatives of sodium-coupled bicarbonate transporters. Physiol Rev. 2013;93:803-959.
- [CrossRef] [PubMed] [Google Scholar]
- Proximal renal tubular acidosis and ocular pathology: A novel missense mutation in the gene (SLC4A4) for sodium bicarbonate cotransporter protein (NBCe1) Mol Vis. 2006;12:324-30.
- [Google Scholar]
- Nephrology and fluid/electrolyte physiology (3rd ed). Amsterdam: Elsevier Health Sciences; 2019.
- [Google Scholar]
- The case | Renal tubular acidosis and eye findings. Kidney Int. 2014;86:217-8.
- [CrossRef] [PubMed] [Google Scholar]
- A novel missense mutation in the sodium bicarbonate cotransporter (NBCe1/SLC4A4) causes proximal tubular acidosis and glaucoma through ion transport defects. J Biol Chem. 2004;279:52238-46.
- [CrossRef] [PubMed] [Google Scholar]
- Mutational and functional analysis of SLC4A4 in a patient with proximal renal tubular acidosis. Pflugers Arch. 2004;448:438-44.
- [CrossRef] [PubMed] [Google Scholar]
- Extrarenal signs of proximal renal tubular acidosis persist in nonacidemic Nbce1b/c-Null mice. J Am Soc Nephrol. 2019;30:979-89.
- [CrossRef] [PubMed] [Google Scholar]




